

A decade’s advice on flow cytometry controls
“A result without an appropriate control is only an anecdote, not an experiment.”
— Modern methodological paraphrase
Dear Researcher,
In last week’s edition, Wildtype One asked Juliano:
What part of spectral flow cytometry do researchers rush and regret later?
He said it’s reference controls:
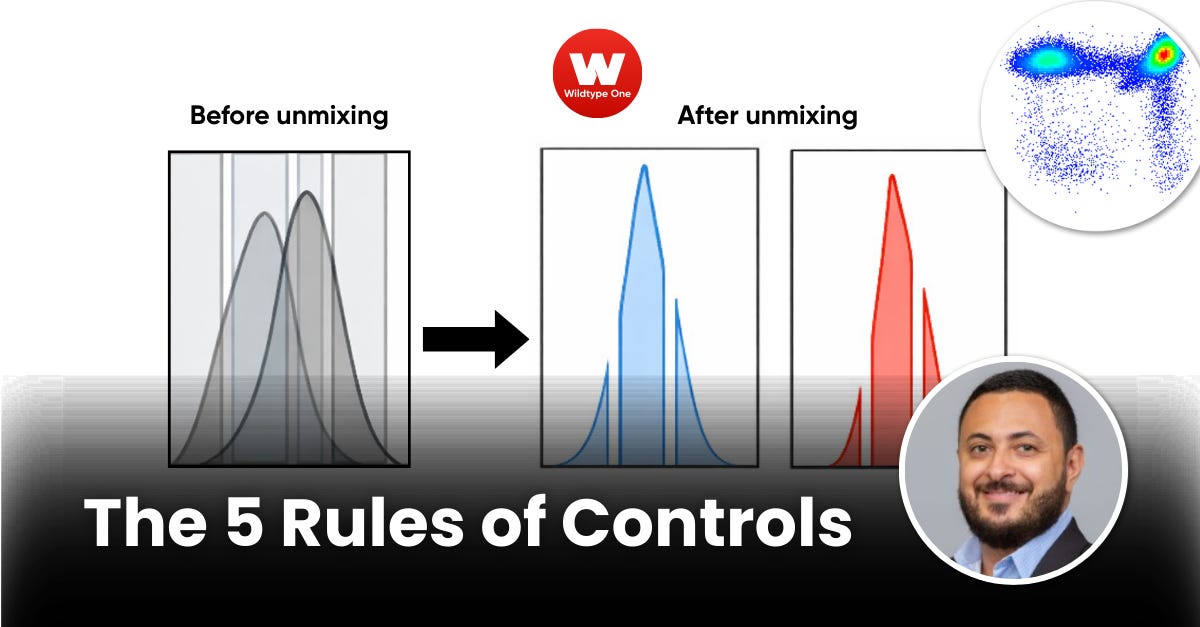
“There are 5 rules. People tried to bend them, and the data punished them for it.”
He has over a decade of experience on flow cytometers and helping researchers generate the best flow data
Here are his rules (in 3 minutes or less):
Rule #1: Record enough events
Yes, the software has a minimum
But that is not what we mean by “enough”
Controls with few events can work
But they’re fragile and noisy
Reliable unmixing means:
Well-sampled,
Positive and negative populations
…that actually represent your biology
Rule:
More events mean cleaner results, and far fewer surprises later.
Rule 2: Clear positive and negative populations
Mushy positives = mushy signatures
Broad/smeared positive population = vague spectral signature
The algorithm is trying to learn what your fluorochrome really looks like
Not average the uncertainty
Rule:
Pick the brightest, cleanest population you can
…and gate it intentionally
Clear positives and clear negatives lead to stable unmixing and fewer downstream headaches.
Rule 3: Positive and negative must share the same autofluorescence
This is where many controls quietly fail
Different cell types, tissues, and treatments come with different autofluorescence profiles
If the positive and negative populations do not share the same autofluorescence, the algorithm cannot cleanly subtract background from signal
The result:
Unstable unmixing,
Increased spread,
and artifacts that look biological until they are not
Rule:
Matching autofluorescence is not a detail; it is a requirement.
Rule 4: Control brightness ≥ Sample Brightness
Controls dimmer than the sample?
The software is then forced to extrapolate beyond what it has seen
That extrapolation is where unmixing errors are born!
Rule:
Bright, well-defined controls give the algorithm confidence
In Juliano’s words:
“Dim controls make it guess—and spectral cytometry does not like guessing.”
Rule 5: The spectrum must be identical
Sounds obvious, yet it’s often broken
Spectral signatures of your reference controls must exactly match the sample
“Similar” is not good enough
“Equivalent” fluorochromes are not equivalent in spectral cytometry
FITC ≠ GFP
PE ≠ tdTomato
APC ≠ AF647
They teach the algorithm the wrong fingerprint, and lead straight to unmixing errors
Tandem dyes also need extra care—their spectra can vary between lots
And don’t forget buffers—fixation and permeabilization can change spectra and autofluorescence
Rule:
Always use the same antibody and the same lot for both controls and samples
And reference controls must be treated exactly like samples
“Yes, even when you use beads as controls.”
Said Juliano
BONUS: The math actually happens during spectral unmixing
Done with acquisition?
Spectral unmixing is what turns raw signals into biological parameters
In Juliano’s words:
“This is the step where spectral cytometry either quietly works or very loudly exposes our mistakes.
“And unlike compensation in conventional flow, you can’t fix this later with a slider and some optimism.
“Unmixing is entirely mathematical; there is no manual rescue mode.”
Unmixing takes the spectral signatures from your reference controls
It then calculates how much each fluorochrome contributed to the total signal measured across all detectors
Every cell starts as a bundle of mixed signals
The raw data contains everything: good signal, spillover, autofluorescence, and all
Unmixing is what separates that mess into individual parameters you can actually analyze.
Clean signatures + solid controls = boring unmixing
…the good kind of boring:
The data behaves
Populations look like biology says they should
You move on with your life
Wrong signatures?
Unmixing gets creative
…the bad kind of creative:
Super negative populations
Weird spreading
Populations that appear convincing until they vanish the moment you fix the controls and re-unmix
These are not exciting biological discoveries; they are math complaining about bad inputs.
Checking unmixing is not optional!
Always look at your reference color controls after unmixing. Always look at your fully stained samples.
If something looks impossible, it probably is. Spectral cytometry is powerful, but it is also very honest.
It will always show you exactly what you taught it.
Spectral flow cytometry is powerful and ruthless
But when those rules are respected, it is one of the most enjoyable ways to do high-parameter cytometry
Controls are real experiments—not a box to check
Trust spectral unmixing
…but always verify what it did before you start interpreting biology
Next advice from Juliano? Spectral panel design: “How to avoid crying in front of your cytometer”
Stay tuned.
As promised, better science in 3 minutes or less.
See you next week,
— Carl from Wildtype One 🧬
PS - Forward this to a colleague to join the newsletter using this link (it’s free).
About Juliano: Juliano Tiburcio de Freitas is a dad, a soccer fan, and an experienced cancer biologist with over a decade of experience operating cytometers. He is a Core Facility Research Specialist at the Cytometry and Imaging Shared Resource at Sylvester Comprehensive Cancer Center, University of Miami, where he supports multicolor and spectral assays, immune profiling, data interpretation, and advanced troubleshooting. He spends his time helping researchers simplify complex flow ideas, avoid issues, and generate good data—and he enjoys it. He earned his PhD from Florida International University, completed postdoctoral training at the University of Miami, with a B.S. from the Federal University of Viçosa in Brazil, and research internship experience in Portugal, Germany, and France. Across his academia and core facility work, flow cytometry has been a constant tool.